



Toward automated classification of bacterial metabarcoding samples by machine learning

Misztak, Agnieszka 
Promotor(s) :
Baurain, Denis
Date of defense : 3-Sep-2021 • Permalink : http://hdl.handle.net/2268.2/12550
 All files (archive ZIP)
All files (archive ZIP) BIM_thesis_AMisztak.pdf
BIM_thesis_AMisztak.pdf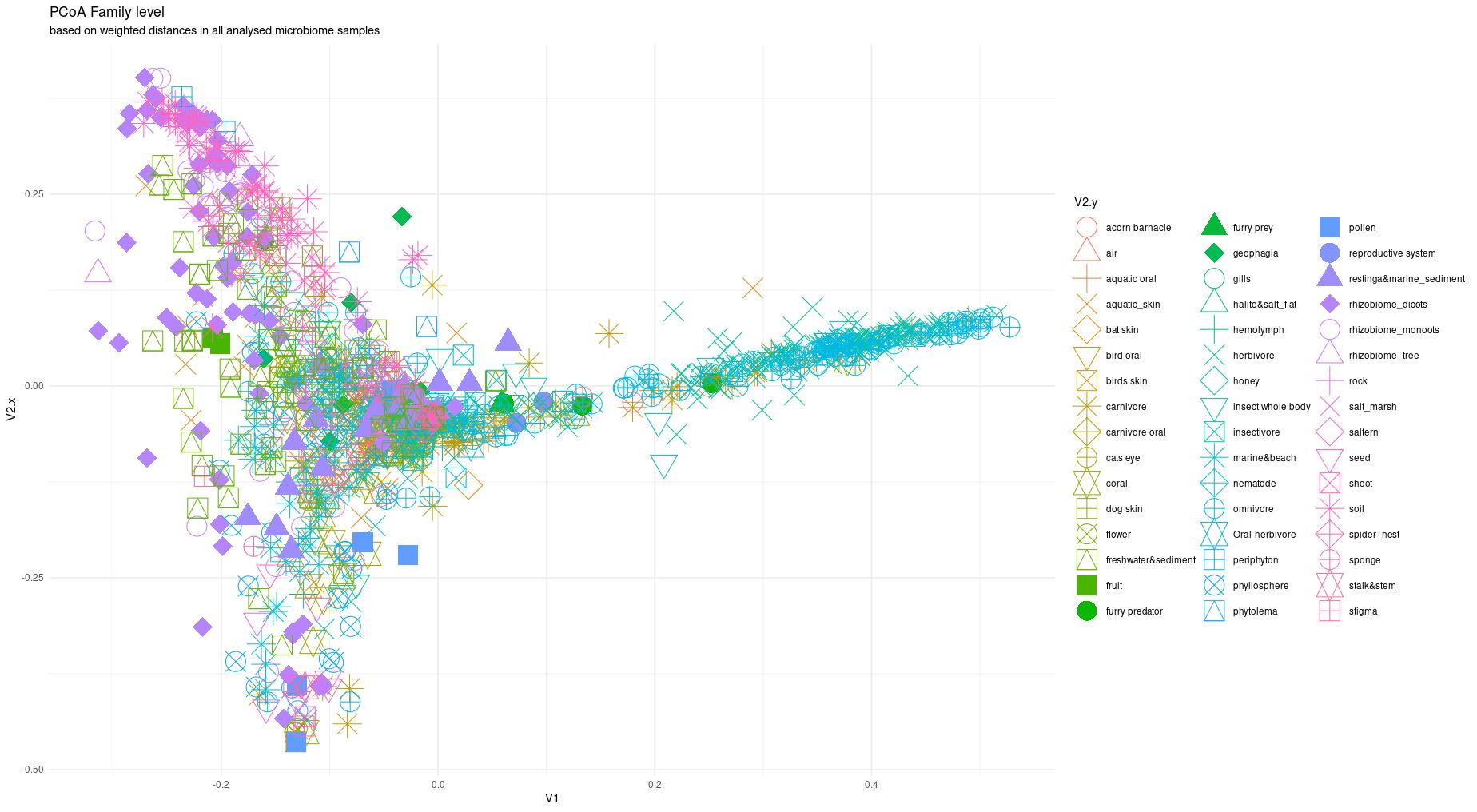 SFig1.jpeg
SFig1.jpeg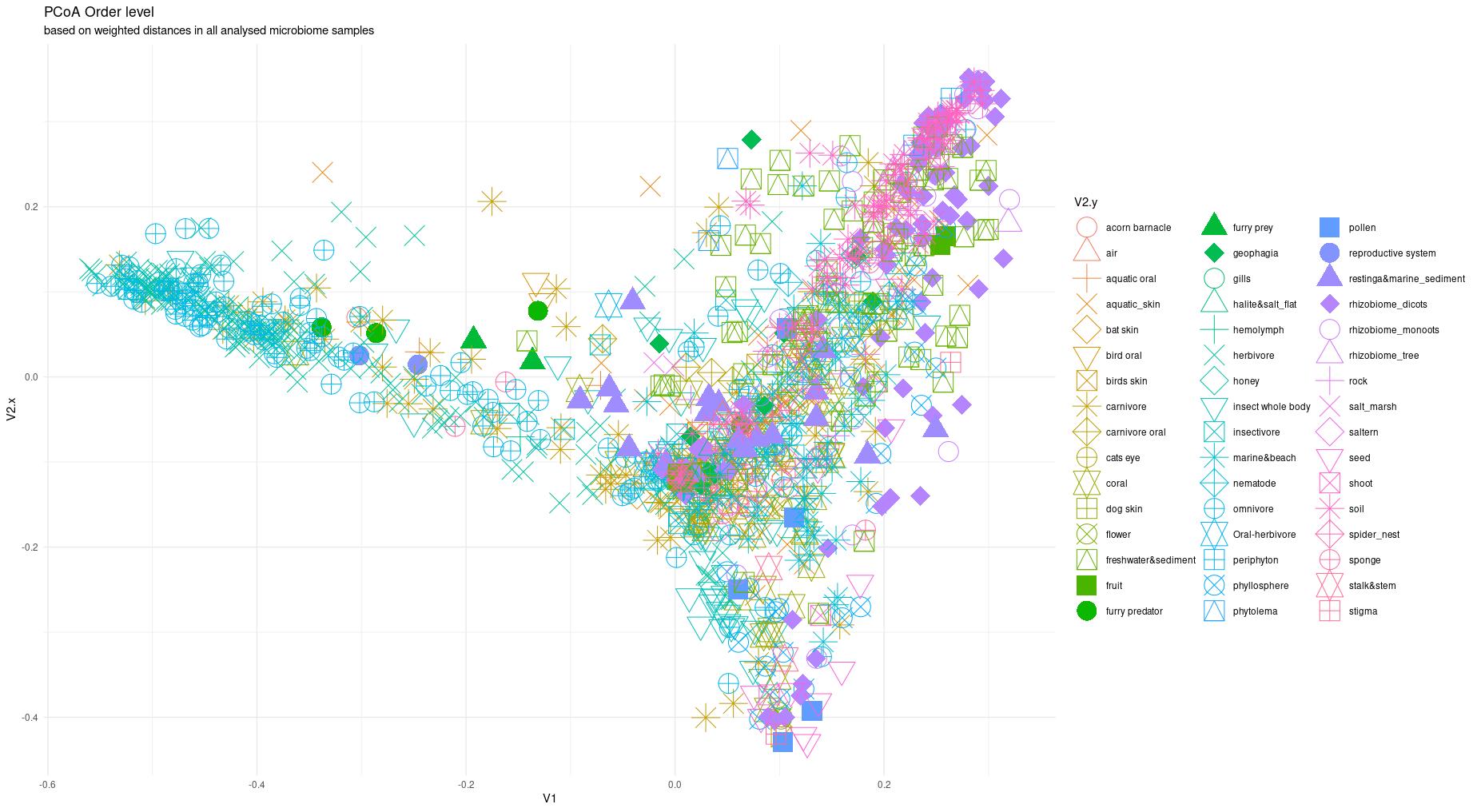 SFig2.jpeg
SFig2.jpeg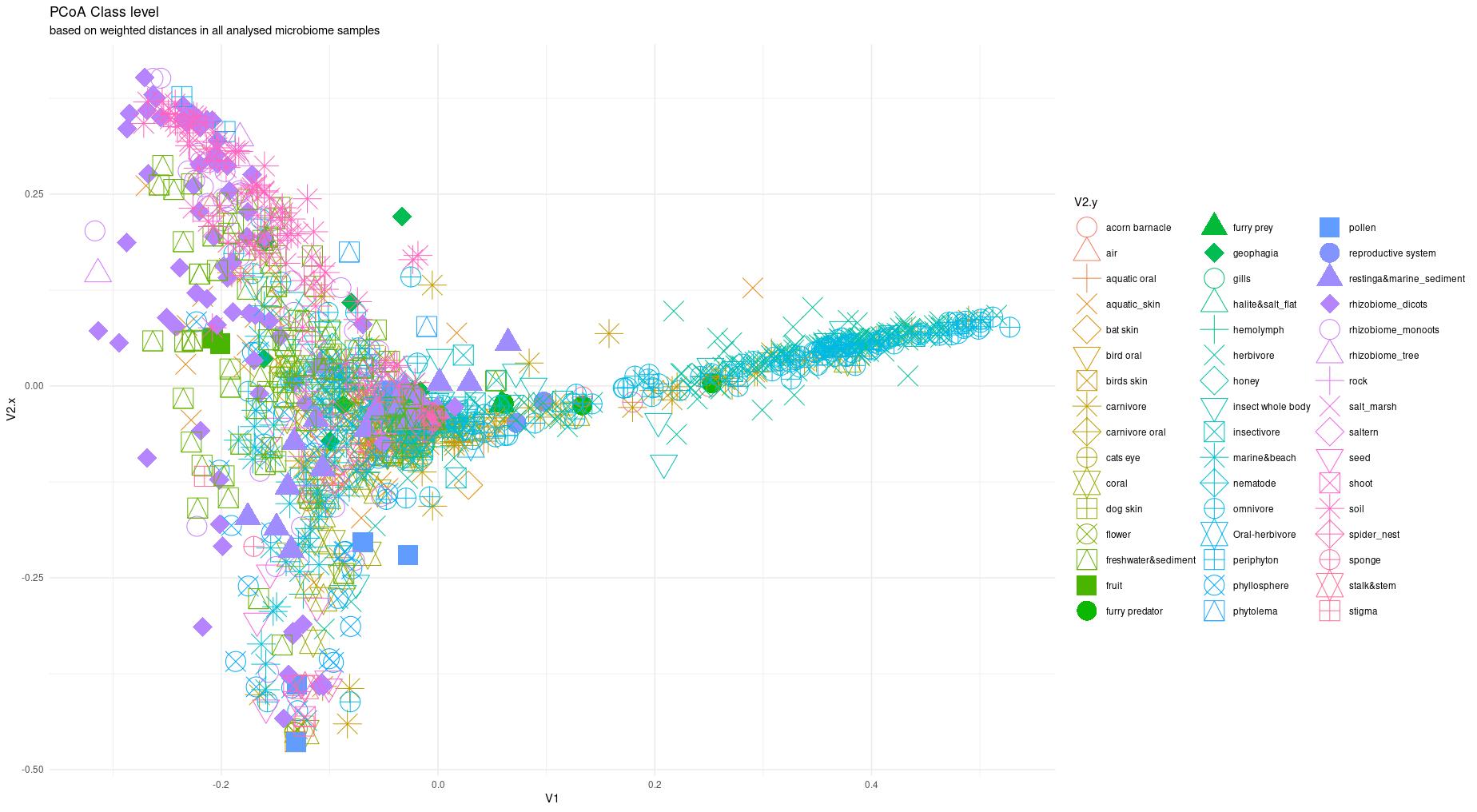 SFig3.jpeg
SFig3.jpeg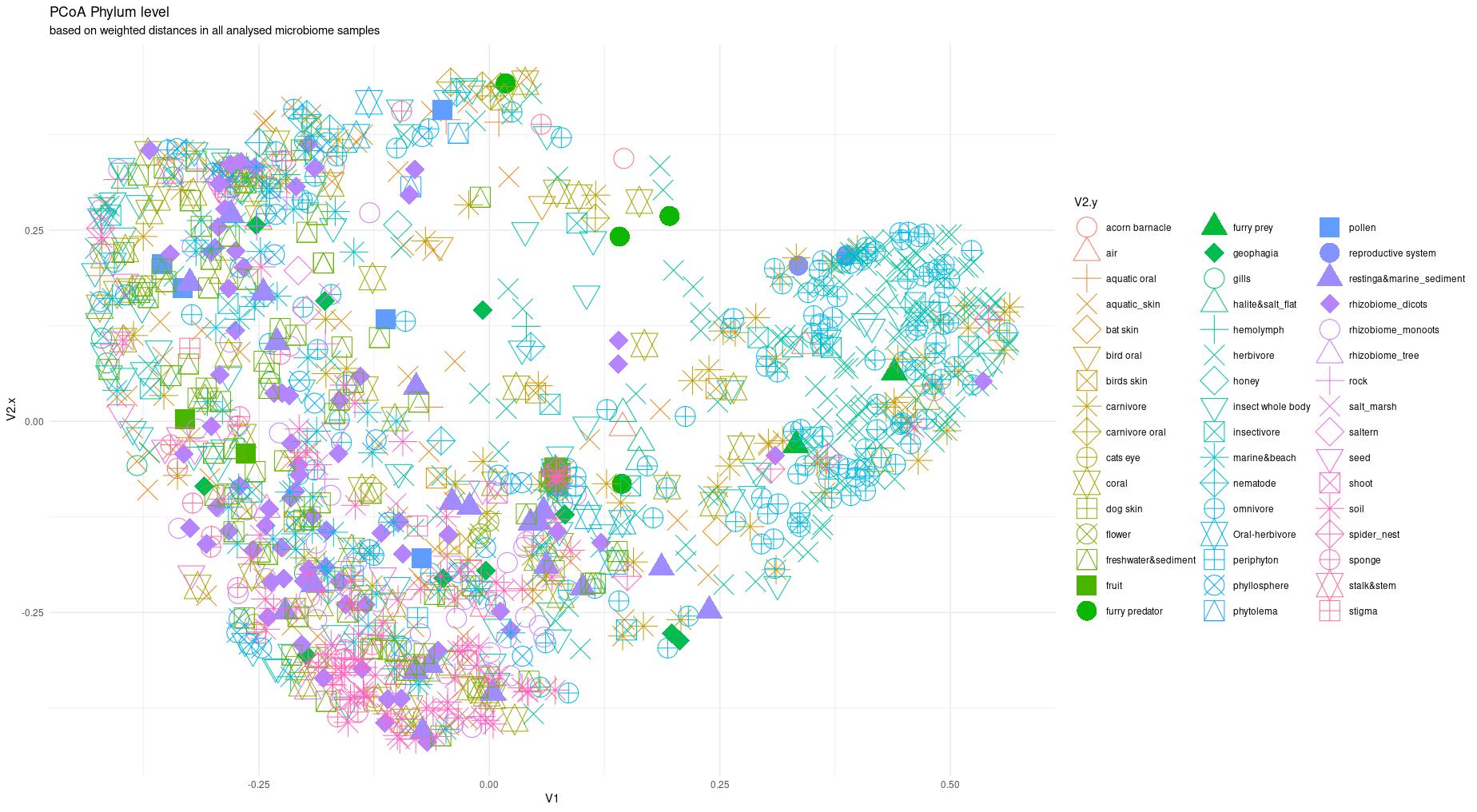 SFig4.jpeg
SFig4.jpeg SFig5.jpeg
SFig5.jpeg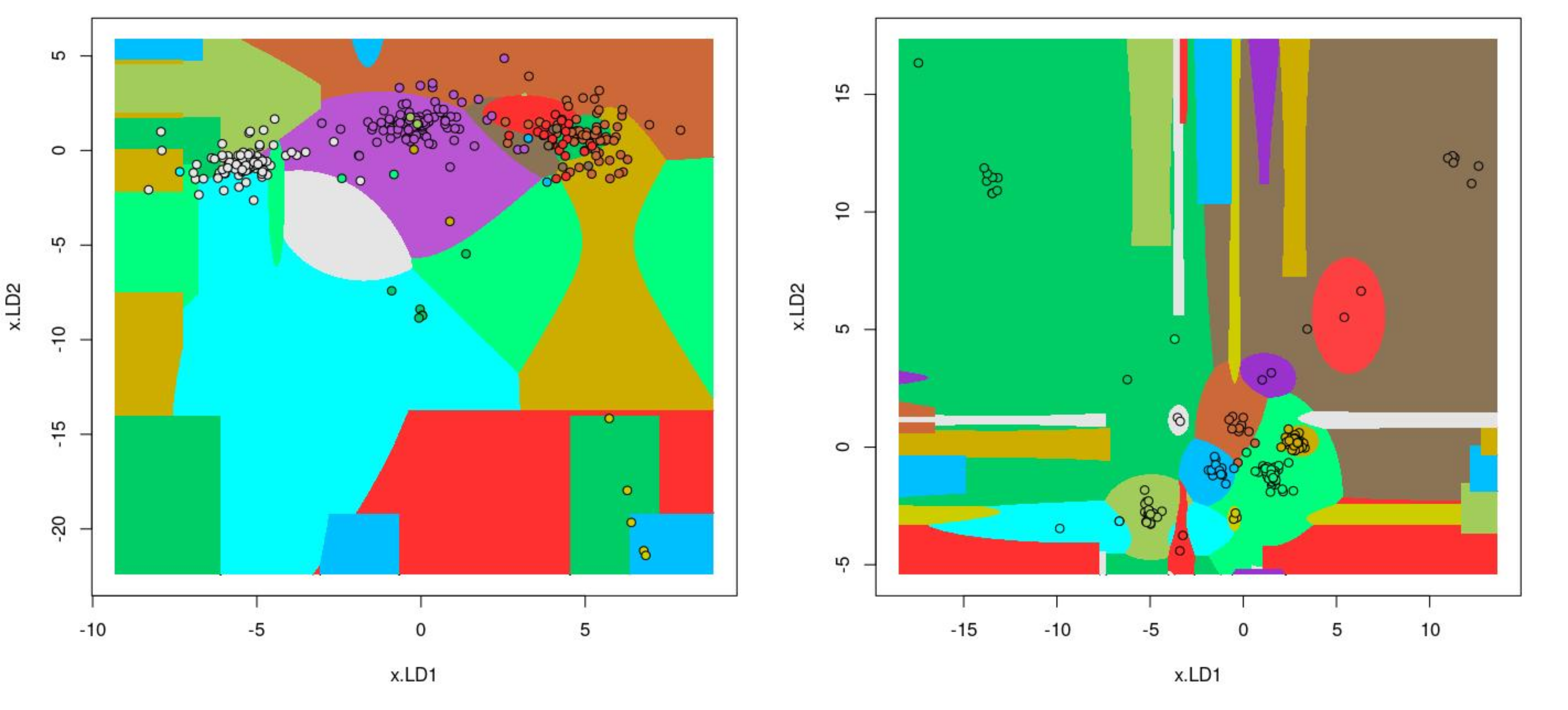 SFig6.png
SFig6.png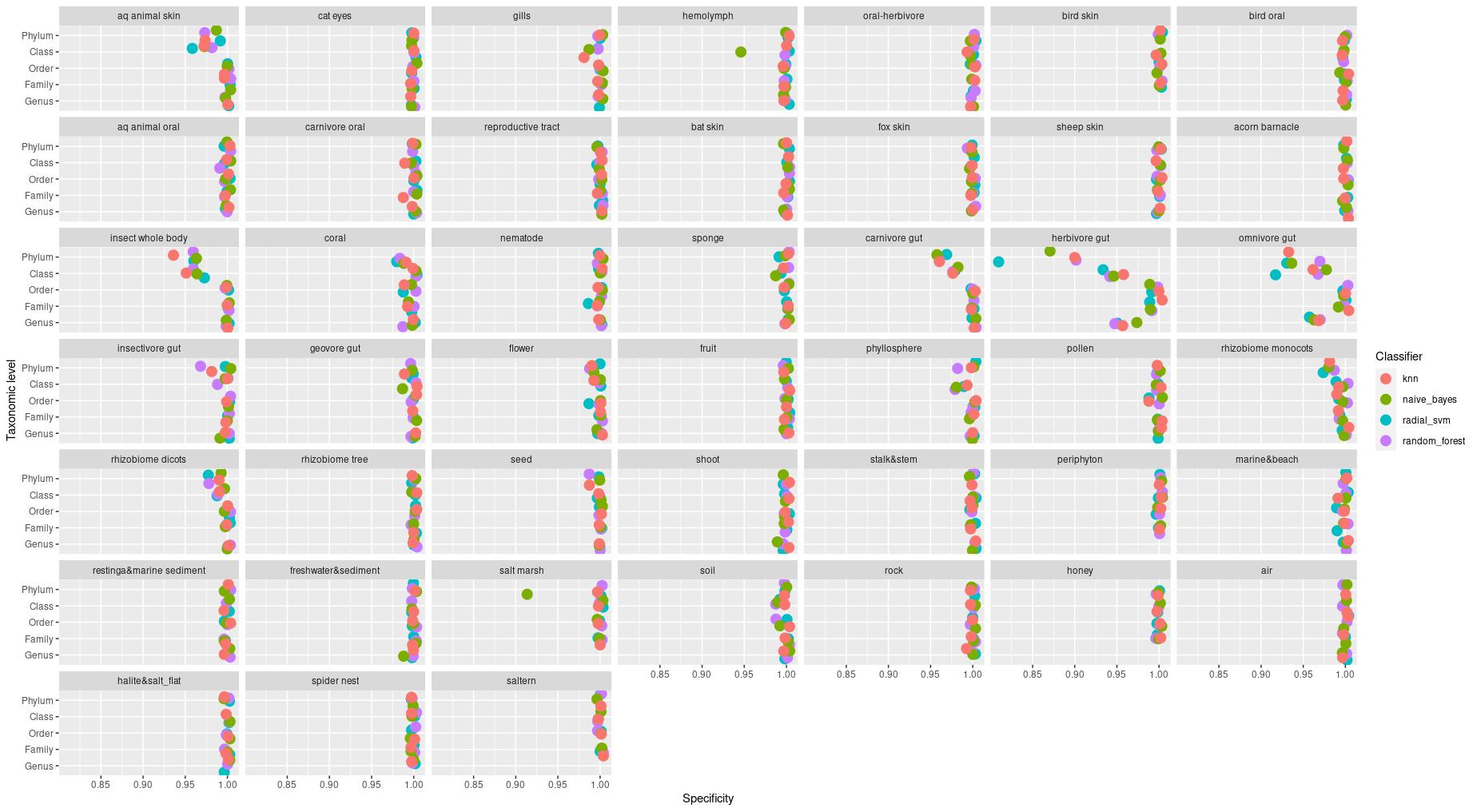 SFig7.jpeg
SFig7.jpeg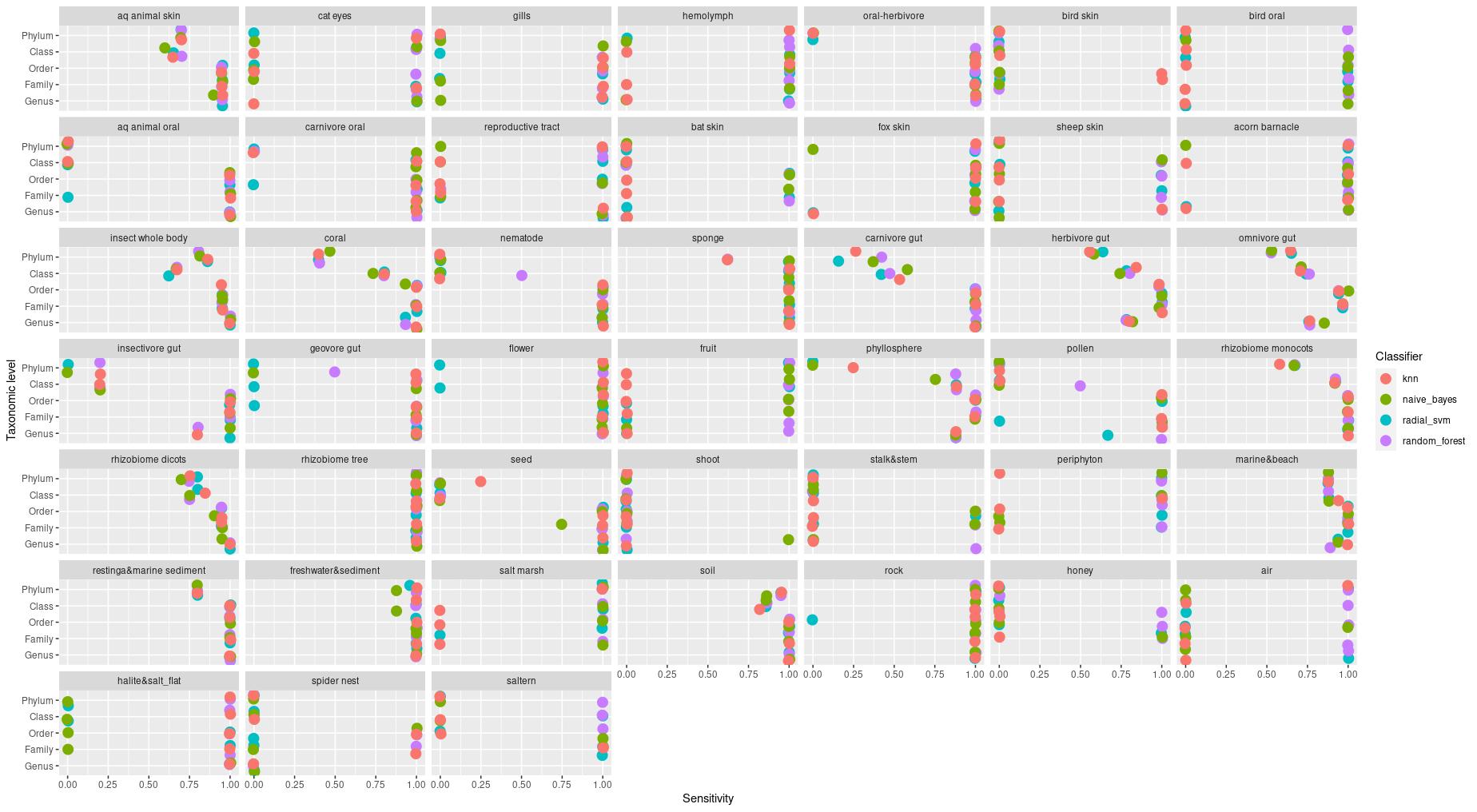 SFig8.jpeg
SFig8.jpeg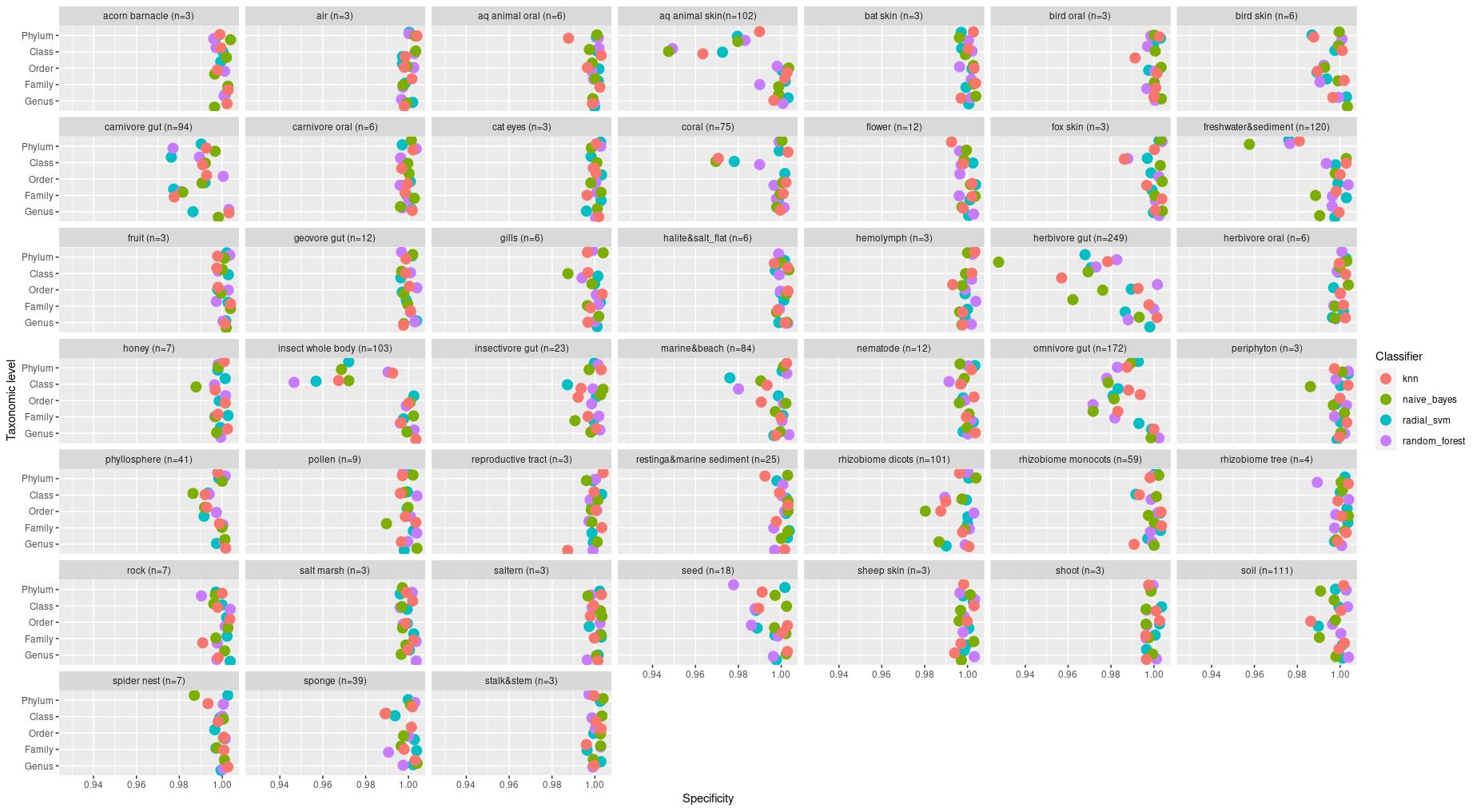 SFig9.jpeg
SFig9.jpegDetails
| Title : | Toward automated classification of bacterial metabarcoding samples by machine learning |
| Author : | Misztak, Agnieszka
|
| Date of defense : | 3-Sep-2021 |
| Advisor(s) : | Baurain, Denis
|
| Committee's member(s) : | Hanikenne, Marc
Meyer, Patrick
Taminiau, Bernard
|
| Language : | English |
| Number of pages : | 48 |
| Discipline(s) : | Life sciences > Microbiology |
| Institution(s) : | Université de Liège, Liège, Belgique |
| Degree: | Master en bioinformatique et modélisation, à finalité approfondie |
| Faculty: | Master thesis of the Faculté des Sciences |
Abstract
[en] The studies of the bacterial communities are increasingly popular. Thanks to the continuous decrease in price of NGS services, curiosity is the limit. It is reflected in the diversity of the metabarcoding data available. Recently a collaborative Earth Microbiome Project had begun a creation of Earth’s multiscale microbial diversity catalogue unifying the effort of almost 100 independent studies for standardization of the protocol for bacterial communities analyses. However, in the public databases there is a substantial amount of the metabarcoding data that were generated throughout the years with the use of different sequencing primers targeting different hypervariable regions.
The information about bacterial communities compositions accumulated in those metabarcoding samples could serve e.g. for identification of the origin of the sample. This work aims at establishing a base process for combining the analysis of the metabarcoding data obtained using various protocols. In the process of selection, out of over a million sequencing runs, 1567 individually processed paired-end reads samples were merged into 45 fine-scaled categories falling into four general datasets: animal-, animal-gut-, environment-, and plant-related. Next, they were processed using popular QIIME2 software without OTU clustering. Three general databases containing 16S rRNA taxonomic information, and their efficacy at five taxonomic ranks, have been tested in order to optimize the taxonomic identification of amplicon sequence variants. The above-mentioned datasets were tested for classification accuracy using two different dimensionality reduction techniques, Principal Component Analysis and Linear Discriminant Analysis applied on the similarity/dissimilarity matrices obtained separetly from an abundance and presence/absence matrices. The aptitude of machine learning in establishing the taxonomic-based classification of the sample sources has been tested with four different algorithms, radial SVM, Naive Bayes, Random Forest and k-Nearest Neighbours. The LDA transformed similarity matrix created at Order rank provided the best and most confident classification with corrected accuracy of 97.6%. Additionally, to examine whether there exist taxonomic relationships among the microorganisms detected in the aforementioned studies, the association rule learning algorithms ‘Apriori’ has been utilized. Number of co-occurrences of microorganisms on different taxonomic ranks was detected and several different taxa forming highly connected nodes were observed. Those taxa can be regarded as putative keystone taxa and considered for further investigation in different niches.
File(s)
Document(s)

Annexe(s)

Cite this master thesis
The University of Liège does not guarantee the scientific quality of these students' works or the accuracy of all the information they contain.
